Crystal Structure of Inhibitor-Bound Bacterial Oligopeptidase B in the Closed State: Similarity and Difference between Protozoan and Bacterial Enzymes.
Petrenko, D.E., Karlinsky, D.M., Gordeeva, V.D., Arapidi, G.P., Britikova, E.V., Britikov, V.V., Nikolaeva, A.Y., Boyko, K.M., Timofeev, V.I., Kuranova, I.P., Mikhailova, A.G., Bocharov, E.V., Rakitina, T.V.(2023) Int J Mol Sci 24
- PubMed: 36768612
- DOI: https://doi.org/10.3390/ijms24032286
- Primary Citation of Related Structures:
7YWP - PubMed Abstract:
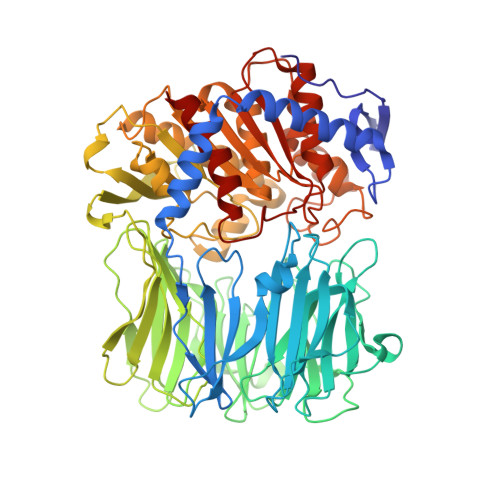
The crystal structure of bacterial oligopeptidase B from Serratia proteamaculans (SpOpB) in complex with a chloromethyl ketone inhibitor was determined at 2.2 ? resolution. SpOpB was crystallized in a closed (catalytically active) conformation. A single inhibitor molecule bound simultaneously to the catalytic residues S532 and H652 mimicked a tetrahedral intermediate of the catalytic reaction. A comparative analysis of the obtained structure and the structure of OpB from Trypanosoma brucei (TbOpB) in a closed conformation showed that in both enzymes, the stabilization of the D-loop (carrying the catalytic D) in a position favorable for the formation of a tetrahedral complex occurs due to interaction with the neighboring loop from the β-propeller. However, the modes of interdomain interactions were significantly different for bacterial and protozoan OpBs. Instead of a salt bridge (as in TbOpB), in SpOpB, a pair of polar residues following the catalytic D617 and a pair of neighboring arginine residues from the β-propeller domain formed complementary oppositely charged surfaces. Bioinformatics analysis and structural modeling show that all bacterial OpBs can be divided into two large groups according to these two modes of D-loop stabilization in closed conformations.
Organizational Affiliation:
National Research Center "Kurchatov Institute", 123182 Moscow, Russia.