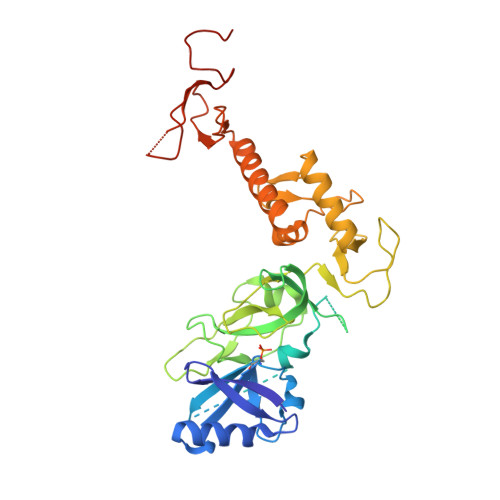
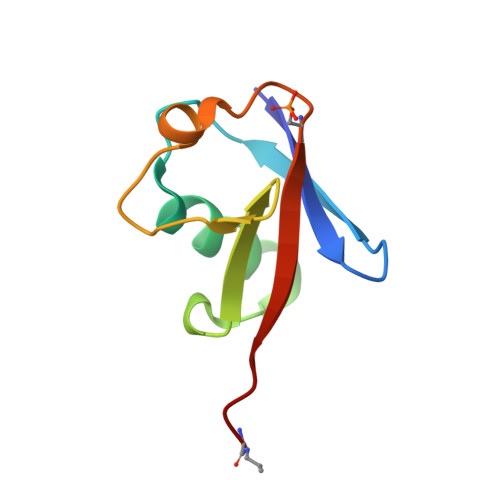
Mechanism of parkin activation by PINK1.
Gladkova, C., Maslen, S.L., Skehel, J.M., Komander, D.(2018) Nature 559: 410-414
- PubMed: 29995846
- DOI: https://doi.org/10.1038/s41586-018-0224-x
- Primary Citation of Related Structures:
6GLC - PubMed Abstract:
Mutations in the E3 ubiquitin ligase parkin (PARK2, also known as PRKN) and the protein kinase PINK1 (also known as PARK6) are linked to autosomal-recessive juvenile parkinsonism (AR-JP) 1,2 ; at the cellular level, these mutations cause defects in mitophagy, the process that organizes the destruction of damaged mitochondria 3,4 . Parkin is autoinhibited, and requires activation by PINK1, which phosphorylates Ser65 in ubiquitin and in the parkin ubiquitin-like (Ubl) domain. Parkin binds phospho-ubiquitin, which enables efficient parkin phosphorylation; however, the enzyme remains autoinhibited with an inaccessible active site 5,6 . It is unclear how phosphorylation of parkin activates the molecule. Here we follow the activation of full-length human parkin by hydrogen-deuterium exchange mass spectrometry, and reveal large-scale domain rearrangement in the activation process, during which the phospho-Ubl rebinds to the parkin core and releases the catalytic RING2 domain. A 1.8?? crystal structure of phosphorylated human parkin reveals the binding site of the phospho-Ubl on the unique parkin domain (UPD), involving a phosphate-binding pocket lined by AR-JP mutations. Notably, a conserved linker region between Ubl and the UPD acts as an activating element (ACT) that contributes to RING2 release by mimicking RING2 interactions on the UPD, explaining further AR-JP mutations. Our data show how autoinhibition in parkin is resolved, and suggest a mechanism for how parkin ubiquitinates its substrates via an untethered RING2 domain. These findings open new avenues for the design of parkin activators for clinical use.
Organizational Affiliation:
Medical Research Council Laboratory of Molecular Biology, Cambridge, UK.